

By: Dr. Farhat Naz (Aurhor)
Dr. Saadia Rehman(Co-author)
Dr. Khazima Asif (Co-author)
Introduction, Aetiology, lnvestigations, Management, Inter professional approach
Case Scenario:
A 25 years old female was admitted in hospital through emergency with complaints of shortness of breath and fatigue which was increasing over the past one week and epistaxis for last one day. The shortness of breath is mostly on exertion and she also complains of dizziness while standing and increasing fatigue. She also had 6-7 episodes of nose bleeds which did not improve by itself.
Past History:
She was born with birth defects, She had abnormal thumb and shoulder but her mobillity was not much affected, She was also hard of hearing and had squint since birth. She was treated as a special child and had delayed developmental milestones and short stature as compared to other siblings. When felt better she took part in housework. She could not recieve formal education in school due to her birth defects and her disease.
She was first came to medical attention in 2014, at 11 years of age when she got hospitalized with similar complaints and her laboratory workup revealed severe anemia. She was diagnosed as a case of Fanconi’s anemia on the basis of her skeletal abnormalities, hematology and bone marrow reports which showed hypo plastic marrow.
She was advised chromosomal analysis at the time of her first bone marrow but record could not be retrieved. Since then; She is being treated through blood transfusions which she needs once or twice monthly.
She has history of multiple blood transfusions and having transfusion once or twice a month for last 11 years. She also had multiple admissions for bleeding and anemia.
Her fresh record and ultrasound done a month ago showed enlarged liver and spleen and hypoechoic areas in liver suggesting metastatic disease.

Family history:
She has no family history of similar illness, Her parents, three brothers and one sister are normal and nobody had anemia and blood transfusions. Her parents had cousin marriage.
According to mother they belong to low socioeconomic background and had to arrange transfusions at their own expense which caused them great economic burden.
Apart from her shortness of breath and fatigue she sometimes developed stomach pain and had black color stool.
On Examination:
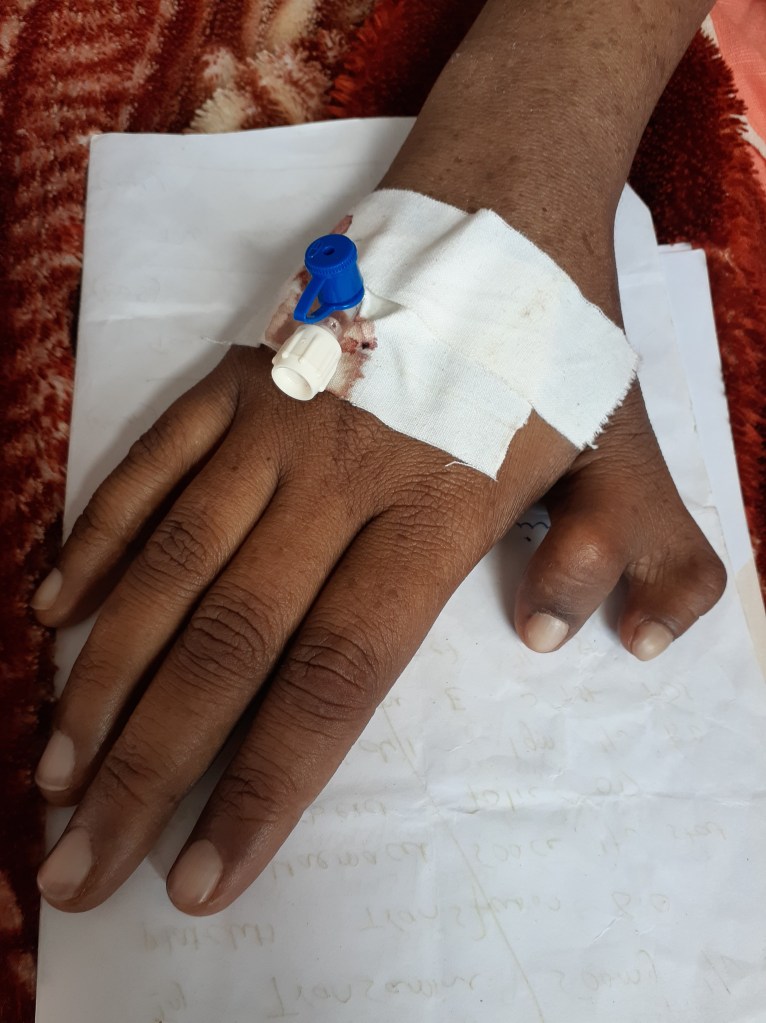
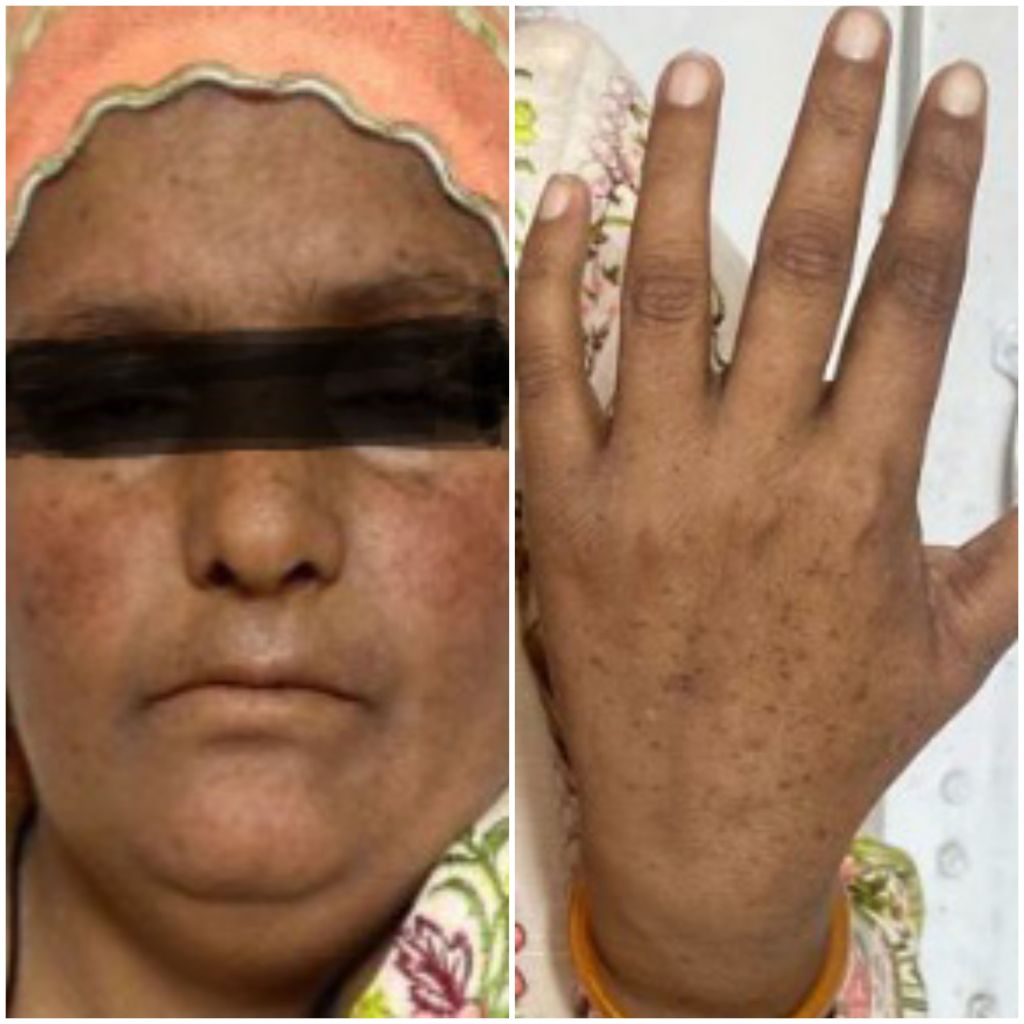
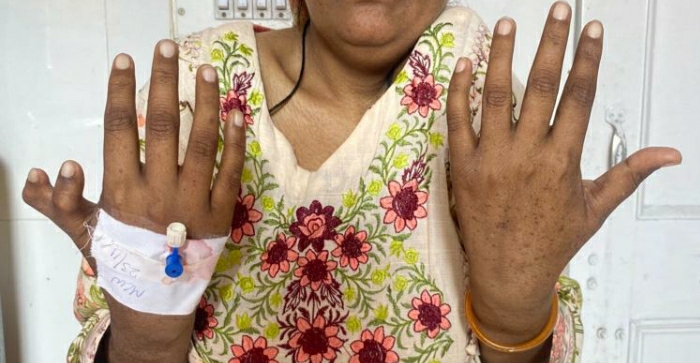
She is a young short stature female with pallor, jaudice, small palpebral fissures, stabismus, microcephaly and pigmentation on face and hands, She has deformed right thumb and absent left clavicle with deformed left shoulder (dysostosis), She is pale and having tinge of jaundice.
She has systolic murmur in heart and mild non tender hepato-splenomegaly. No lymph nodes were palpable.


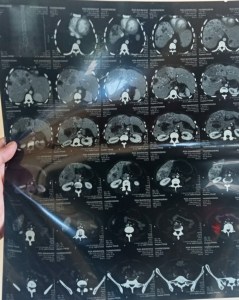
Radiology:
CT scan of abdomen showing multiple hypoechoic areas suggestive of metastatic disease involving liver and spleen.
Laboratory Findings:
Hb: 5.9g/dl
Hematocrit: 25%(38-52%)
RBC Count:3.8×10⁶/micro litre(4.5-5.5×10⁶/ micro litre
WBC Count: 1.7×10³/ microlitre (4.5-11×10³ microlitre with lymphocytosis (85%)
Platelet count: 30×10³/ microlitre (150-300×10³/microlitre)
MCV: 111
Total Bilirubin: 3.7 mg/dl (0.1-1.0)
Direct Bilirubin: 1.9 mg/dl
Indirect Bilirubin: 1.8mg/dl (0-0.84)
ALT: 208 U/l (10-50)
ALP: 328 U/L (30-120)
Serum Ferritin: 11195 (high) (23.9-336.2)
LDH: 324 U/L (<248)
PT: 33 (control 16)
INR: 1.7
Albumin: 4g/dl (3.5-5.5)
Renal Function Tests: Normal
Electrolytes: Normal
Microscopic Examination:
Peripheral Blood Smear:The peripheral blood smear revealed:
Anisocytosis(Red blood cells exhibit significant size variation, ranging from microcytes to macrocytes)
Poikilocytosis: (Abnormal red cell shapes are observed, including elliptocytes, teardrop cells, and target cells)
Hypochromia: (Many red blood cells appear pale, indicating reduced hemoglobin content)
Leukopenia: (The white blood cell count was decreased, with decreased numbers of granulocytes, lymphocytes, and monocytes.
Thrombocytopenia: (The platelet count was reduced, and several giant platelets were seen).
Bone Marrow Trephine Biopsy:
Hypoplastic marrow with depressed all the three cell lines, bone marrow mostly replaced by adipose tissue.
Follow up:
She was followed and got admitted twice with bleeding. Her contrast enhanced CT of abdomen done later showed extensive metastasis in liver and spleen. Her chest wall was deformed and lungs had ground glass opacification. Her pelvic scan was normal. No immature cells were identified in peripheral smear. Her extensive work up could not find the primary lesion of her extensive metastasis.
She developed bleeding and profuse diarrhoea and developed septicemia from which she could not revive.
- What is Fanconi’s anemia?
- How Fanconi’s anemia is inherited and the disease is caused?
- What are the presenting symptoms of Fanconi’s anemia?
- What are the clinical features of Fanconi’s anemia?
- What are the anemia-related symptoms of Fanconi’s anemia?
- What are the skeletal abnormalities associated with Fanconi’s anemia?
- What are the cardiovascular abnormalities associated with Fanconi’s anemia?
- What are the skin abnormalities associated with Fanconi’s anemia?
- What are the complications of Fanconi’s anemia?
- What investigations are done to diagnose Fanconi’s anemia?
- What is the management and prognosis of Fanconi’s anemia?
- What is the role of stem cell transplantation in treating Fanconi’s anemia?
- What is the life expectancy of patients with Fanconi’s anemia
What is Fanconi’s anemia?
Fanconi anemia (FA) is a rare genetic disorder characterized by bone marrow failure, congenital malformations, and an increased risk of cancer.
It is inherited in an autosomal recessive manner, meaning that an affected individual must inherit two copies of the mutated gene – one from each parent.
prevalence of 1 in 160,000-300,000 individuals globally, but its incidence varies among different populations.
How Fanconi’s anemia is inherited?
There are several modes of inheritance in Fanconi’s anemia and most of the times, it is autosomal recessive but as almost 23 genes have been identified with mutations causing Fanconi’s anemia, it may present with other modes of inheritance including X-linked recessive and rarely in autosomal dominant pattern.
What are the presenting symptoms of Fanconi’s anemia?
Symptoms of Fanconi anemia usually appear during childhood, and they may include developmental delays, small stature, abnormal skin pigmentation, skeletal abnormalities, kidney problems, hearing loss, and abnormal facial features.
FA patients have an increased risk of developing several types of cancer, including acute myeloid leukemia (AML), squamous cell carcinoma, and other solid tumors.
What are the clinical features of Fanconi’s anemia?
FA patients may present with various clinical features, including anemia, skeletal abnormalities, cardiovascular abnormalities, skin abnormalities, and an increased risk of certain cancers. Anemia-related symptoms of FA can include fatigue, weakness, and shortness of breath. Skeletal abnormalities associated with FA can include short stature, malformed bones in the arms and legs, and scoliosis. Cardiovascular abnormalities associated with FA can include congenital heart defects and an increased risk of heart disease.
Skin abnormalities associated with FA can include hyperpigmentation, café- au-lait spots, and abnormal skin pigmentation.
The frequencies of different defects is as follows:
- Bone marrow defects: 90%
- Birth defects: 75%
- Short stature: 50%
- Skin pigmentation 50%
What is the pathogenesis of Fanconi’s anemia?
The clinical features of FA are caused by a defect in DNA repair mechanis, which leads to chromosomal instability and impaired cell division. FA patients exhibit hypersensitivity to DNA-damaging agents such as mitomycin C and diepoxybutane.
The underlying molecular defects in FA involve at least 22 genes, which are involved in the FA/BRCA DNA repair pathway.
Mutations in these genes can cause defects in DNA repair, resulting in chromosomal instability and results in bone marrow failure and increased cancer susceptibility.
What are the anemia-related symptoms of Fanconi’s anemia?
Anemia-related symptoms of Fanconi’s anemia can include fatigue, weakness, and shortness of breath. The patient may present with fever due to low white cell count and bleeding, petechiae and bruising due to low platelets.
What are the skeletal abnormalities associated with Fanconi’s anemia?
Skeletal abnormalities associated with Fanconi’s anemia can include short stature, malformed bones in the arms and legs, and scoliosis.
Upper Limb:
Thumb: May be bifid, absent, short or supernumerary.
Radius and ulna: Dysplastic or absent.
Lower Limb:
polydactyly, short toes, club feet, flat feet, abnormal femur, hip dislocation, thigh osteoma.
head and face anomalies:
Microcephaly, hydrocephalus
Frontal bossing, flat head
Micrognathia
Webbed short neck, low hairline
Axial: Spina bifida, scoliosis, abnormal ribs, extra vertebrae
Eyes: Epicanthal folds, proptosis, ptosis, cataract blindness, epiphora
Ears: absent ear drums, small or large pinnae, atresia of external auditory meatus,
Gastrointestinal: Imperforate anus, tracheoesophageal fistula, atresia of intestine, Meckel’s diverticulum, megacolon, umbilical hernia.
Renal: Malformations of kidney and urinary tract.
Other: Hypogonadism in both male and female.
What are the cardiovascular abnormalities associated with Fanconi’s anemia?
Cardiovascular abnormalities associated with Fanconi’s anemia can include congenital heart defects like patent ductus arteriosus (PDA), Atrial and Ventricular septal defects (ASD/ VSD) and an increased risk of acquired heart diseases
What are the skin abnormalities associated with Fanconi’s anemia?
Skin abnormalities associated with Fanconi’s anemia can include hyperpigmentation, café-au-lait spots, and abnormal skin hyper pigmentation. The areas of hyperpigmentation mostly include face, trunk, neck, armpits and groin.
What are the complications of Fanconi’s anemia?
The complications of Fanconi’s anemia can include leukemia and other cancers, bone marrow failure, and organ damage.
FA patients are at increased risk for several types of cancer, with the most common being Acute Myeloid Leukemia. Other cancers that can occur in FA patients include solid tumors such as head and neck squamous cell carcinoma and liver tumors. The incidence of cancer in FA patients varies among different populations, but it is estimated that approximately 30-40% of FA patients develop cancer by the age of 40.
What investigations are used to diagnose Fanconi’s anemia?
Diagnosis is usually delayed till the development of aplastic anemia. The average age of diagnosis is 7 years. Diagnosis of FA is based on a combination of clinical features, genetic testing, and laboratory tests.
Laboratory tests:
Complete Blood Count:
It typically shows low Hemoglobin and decreased Red blood cells, white blood cells and platelets.
Serum Erythropoietin: is increased due to low hemoglobin.
Bone marrow biopsy: Bone marrow aspiration shows dry tap and biopsy shows hypoplastic marrow and fatty infilteration.
Genetic testing:
The diagnosis is typically confirmed through genetic testing, which can identify mutations in one of the 22 FA/BRCA genes.
Chromosomal breakage test: Diagnostic test indicated in severe pancytopenia. DNA cross linkers such as Diepoxybutane or Mitomycin stimulates the breakage of DNA in the absence of DNA repair system. Cultured fibroblasts or skin fibroblasts are used to demonstrate chromosomal fragility.
Flow Cytometry: Cell cycle analysis flow cytometry is an alternative to chromosomal breakage test. In this test, those cells with impaired DNA repair undergo G2 arrest following exposure to DNA cross-linking agent.
Fanconi’s Anemia Gene sequencing: is recommended in those showing positive chromosomal breakage to exclude other conditions causing chromosomal breakage.
Prenatal testing: Chromosomal breakage can be demonstated in amniotic fluid cells or on Chorionic Villous Biopsy while raised serum alpha protein is rapid screening test with low sensitivity.
Skeletal survey and Imaging:
Skeletal survey is done to identify bone defects.
X-rays: X-rays are done to know the site and extent of skeletal defects.
CT/ MRI’s: these are done to diagnose CNS abnormalities like absent corpus callosum, hypoplastic cerebellum and small pituitary.
Ultrasound Abdomen: It is done to diagnose kidney and liver abnormalities and to know about malignancy.
What is the management and prognosis of Fanconi’s anemia?
The management of FA includes regular monitoring, blood transfusions, and the use of growth factors to promote the production of blood cells. The use of Stem Cell Transplantation (SCT) is a potential cure for FA and can restore normal blood cell production in patients with the disorder. SCT has been shown to improve survival rates in FA patients, particularly in those who receive the transplant before the onset of severe complications.
Prognosis:
The prognosis of FA varies, but it depends on the severity of the disease and the presence of complications such as bone marrow failure and cancer. With proper management, some FA patients can survive into adulthood. However, the use of SCT is associated with significantly improved survival rates, and it is considered the most effective treatment for FA.
What is the role of stem cell transplantation in treating Fanconi’s anemia?
Stem cell transplantation (SCT) is a potential cure for Fanconi’s anemia and can restore normal blood cell production in patients with the disorder.
What is the life expectancy of patients with Fanconi’s anemia without stem cells transplantation?
The life expectancy of patients with Fanconi’s anemia without a bone marrow transplant varies, but many patients do not survive beyond their 30s or 40s due to complications associated with the disorder.
What is the role of interdisciplinary approach in Fanconi’s anemia?
The care of a patient with Fanconi’s anemia is better if an inter-professional approach is applied in management. The following specialities can join hands to diagnose/manage the disease and monitor /delay the complications. Though mainly a hematological disorder but the following specialities can play the role in managemen
- A haematologist
- A general physician
- oncologist
- Gynaecologist
- Radiologist
- Oncologist
- Pathologist, gene analysts
- Physiotherapists
- Pharmacists
- Nurses
- Friends and family members
Summary:
In conclusion, FA is a rare genetic disorder characterized by bone marrow failure, congenital malformations, and an increased risk of cancer. Diagnosis is typically based on a combination of clinical features, genetic testing, and laboratory tests. Management includes regular monitoring, blood transfusions, and the use of growth factors, while SCT is a potential cure for FA and can restore normal blood cell production. The prognosis of FA varies, but SCT is associated with significantly improved survival rates.
